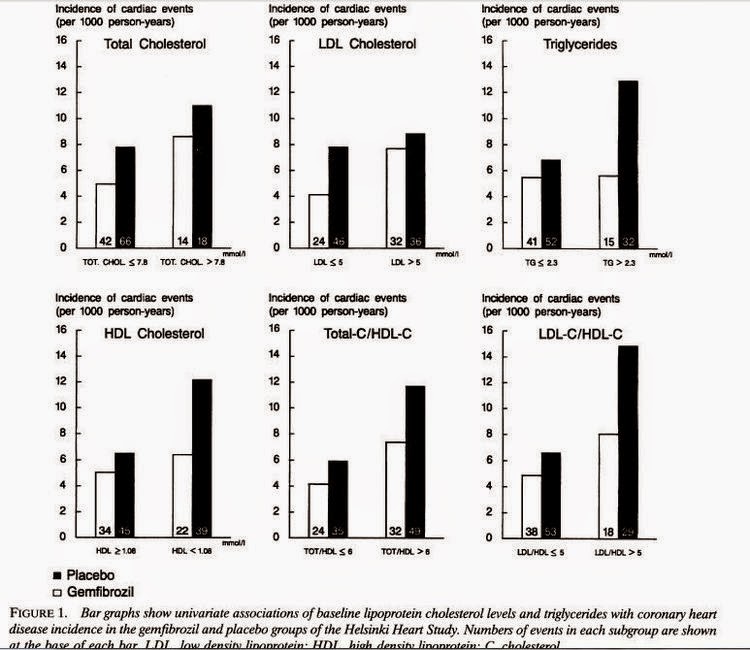
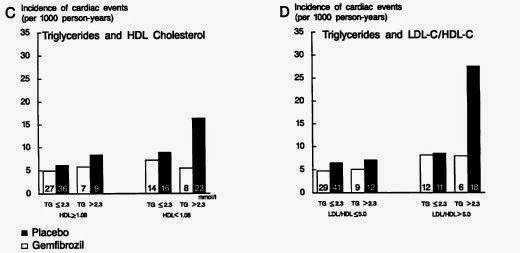
This table is from Dr Malcolm Kendrick's latest blog post, which is about the possibility of a retrospective "publication bias" deleting findings that don't support the lipid hypothesis on certain websites. The data, from the European Heart Study 2008, has been tabulated by Dr Kendrick to show the correlation between saturated fat consumption and CHD mortality between countries.
![guessinggame]()

What's striking is the big gap between the countries of the former Soviet Union and the Western European states (and, for some reason, Israel). France, with the lowest CHD mortality, has the world's highest per capita butter consumption, Switzerland is similar, and olive oil countries don't come out too badly either. The UK, with its chicken twizzlers, mars bars, and fish fingers, and Israel, with its combination of soy oil plus high tech medical care, don't come out quite so well.
The massive rate of CHD in former Soviet states is attributable to many things - industrial and agricultural pollution, smoking and alcohol, untreated chronic infections, overwork, malnutrition, higher birthrate, lower incomes (the US CHD rate in 2008 was 126 per 100,000 - this is both sexes, age adjusted, so not exactly comparable to Dr Kendrick's table; see here for more age-adjusted data and discussion). To look at the correlations between these things and CHD is enough to question the existence of any diet-heart link whatsoever. What significance does a RR of 1.17 have in a world where RRs of 11.13 exist?
If, however, we must look at these statistics in diet-heart terms, one thing stands out to this student of history. Russia is the world's oldest producer, and consumer of vegetable seed oils. The sunflower was brought to Europe by the Spanish around 1510, and were established in the Netherlands (then part of the Spanish Empire) soon after. Peter the Great then brought the sunflower to Russia after his visit to the Netherlands in 1698. In 1716 a patent was granted in Great Britain for a method of extracting oil from sunflower seeds, and during the 1840s the Tsarist government of Russia began the manufacture of sunflower oil on an industrial scale.
Because the Lent restrictions of the Russian Orthodox Church forbade the consumption of fat, this seemed like a good idea at the time (once again the ascetic impulse will be the driver for a dietary change later to be justified and entrenched by theories about health).
The Great Soviet Encyclopaedia of 1979 naturally downplays the Tsarist achievement.
And so on - the communist love of boring statistics was useful after all.
But wait - there's more. If the Tsars boosted the seed oil industry, the Bolsheviks, for political reasons, destroyed whatever dairy industry there was in Russia during their genocidal campaign against the "kulaks", which they defined as any farmer rich enough to own a cow, plus anyone they didn't like or who opposed their seizures of food, summary executions, and so on.
To destroy the dairy farmers they needed a substitute - so Soviet Russia, beginning in the 1920s, became the first large scale producer of soy products. The USSR was the first nation in Europe and the second nation in the Western world (after the USA) to become a major producer of soybeans. Soybean production, which reached significant levels in the mid-1920s, rose to a remarkable peak of 283,000 tonnes in 1931, but had fallen back to a low of 54,000 tonnes in 1935, after which it increased steadily. At the time of this peak, starting in 1931, the USSR built a large Soybean Research Institute in Moscow, attracted some of the top soybean and soyfoods researchers from western Europe (Rouest, Berczeller), and did extensive soyfoods research, focusing on soymilk and tofu, during the early 1930s.
So by any utopian diet-heart, lipid hypothesis theory of history, those former Soviet states should have had CDH beat years ago.
By the test of reality, on the other hand, you would be better off living in France on butter, cheese, cream and, hey, if you like it why not, olive oil.
The massive rate of CHD in former Soviet states is attributable to many things - industrial and agricultural pollution, smoking and alcohol, untreated chronic infections, overwork, malnutrition, higher birthrate, lower incomes (the US CHD rate in 2008 was 126 per 100,000 - this is both sexes, age adjusted, so not exactly comparable to Dr Kendrick's table; see here for more age-adjusted data and discussion). To look at the correlations between these things and CHD is enough to question the existence of any diet-heart link whatsoever. What significance does a RR of 1.17 have in a world where RRs of 11.13 exist?
If, however, we must look at these statistics in diet-heart terms, one thing stands out to this student of history. Russia is the world's oldest producer, and consumer of vegetable seed oils. The sunflower was brought to Europe by the Spanish around 1510, and were established in the Netherlands (then part of the Spanish Empire) soon after. Peter the Great then brought the sunflower to Russia after his visit to the Netherlands in 1698. In 1716 a patent was granted in Great Britain for a method of extracting oil from sunflower seeds, and during the 1840s the Tsarist government of Russia began the manufacture of sunflower oil on an industrial scale.
Because the Lent restrictions of the Russian Orthodox Church forbade the consumption of fat, this seemed like a good idea at the time (once again the ascetic impulse will be the driver for a dietary change later to be justified and entrenched by theories about health).
The Great Soviet Encyclopaedia of 1979 naturally downplays the Tsarist achievement.
There were about 10,000 small vegetable oil and fat
production shops and about 400 licensed, poorly equipped oil and fat plants in
tsarist Russia. The vegetable oil output in 1913 was 538,000 tons; in addition,
the equivalent of 192,000 tons of soap was produced (figured at a 40-percent
fatty-acid content).
Under Soviet power, the vegetable oil and fat industry has
become one of the major sectors of the food-processing industry, relying on
advanced technology and a stable raw materials base. There are enterprises of
the vegetable oil and fat industry in all of the Union republics. The largest
are combines in Krasnodar, Moscow, Tashkent, Dushanbe, Irkutsk, Saratov, Kirovabad,
Sverdlovsk, Gomel’, and Kazan, which account for 45 percent of the USSR’s total
output of vegetable oil, about 65 percent of its margarine, and more than 75
percent of its soap and detergents.
In 1972 the vegetable oil and fat industry accounted for 5.4
percent of the gross output of the food-processing industry of the USSR, 2.5
percent of the work force, and 2.7 percent of the fixed industrial production
assets.
The USSR is the world’s second largest producer of vegetable
oils, soap, and margarine (after the USA). It accounts for more than 14 percent
of the world’s vegetable oil. The output of vegetable oil in the USSR is
growing steadily; production in 1972 was 3.6 times that of 1940 (see Table 1).
Owing to the increase in agricultural production, state
purchases of oil-yielding crops in 1972 were twice the 1940 figure. The oil
content of sunflower seeds, which account for 50 percent of all seeds processed
by industry, has risen significantly. The material and technical basis for the
vegetable oil and fat industry has grown. Production capacities for processing
oil-yielding seeds have been increased primarily by modernizing existing
extraction plants and building new ones. Introduction of the extraction method
of processing oil-yielding seeds has made it possible to increase labor
productivity, mechanize and automate production processes, and sharply increase
the oil output from raw materials (see Table 2).
The proportion of oil-yielding raw materials processed by
progressive extraction methods increased from 9.9 percent in 1940 to 81 percent
in 1972.
Production in the margarine and soap industries is fully
mechanized.
In the other socialist countries the vegetable oil and fat
industry is based primarily on local raw materials. The volume of production
has generally satisfied the needs of these countries. In 1972 the vegetable oil
output in Rumania was 360,000 tons; in Poland, 213,000 tons; in Yugoslavia,
165,000 tons; in Bulgaria, 145,000 tons; in the German Democratic Republic,
131,-000 tons; in Czechoslovakia, 88,000 tons; and in Hungary, 80,000 tons.
The production of vegetable oil in certain capitalist
countries was as follows: 830,000 tons in Italy (1972), 801,000 tons in the
Federal Republic of Germany (1971), and 520,000 tons in France (1971). In the
USA vegetable oil production in 1972 came to 4.6 million tons; the output of
margarine was 2.6 million tons, and that of soap and synthetic detergents was
3.5 million tons.
And so on - the communist love of boring statistics was useful after all.
But wait - there's more. If the Tsars boosted the seed oil industry, the Bolsheviks, for political reasons, destroyed whatever dairy industry there was in Russia during their genocidal campaign against the "kulaks", which they defined as any farmer rich enough to own a cow, plus anyone they didn't like or who opposed their seizures of food, summary executions, and so on.
To destroy the dairy farmers they needed a substitute - so Soviet Russia, beginning in the 1920s, became the first large scale producer of soy products. The USSR was the first nation in Europe and the second nation in the Western world (after the USA) to become a major producer of soybeans. Soybean production, which reached significant levels in the mid-1920s, rose to a remarkable peak of 283,000 tonnes in 1931, but had fallen back to a low of 54,000 tonnes in 1935, after which it increased steadily. At the time of this peak, starting in 1931, the USSR built a large Soybean Research Institute in Moscow, attracted some of the top soybean and soyfoods researchers from western Europe (Rouest, Berczeller), and did extensive soyfoods research, focusing on soymilk and tofu, during the early 1930s.
So by any utopian diet-heart, lipid hypothesis theory of history, those former Soviet states should have had CDH beat years ago.
By the test of reality, on the other hand, you would be better off living in France on butter, cheese, cream and, hey, if you like it why not, olive oil.